Esophageal atresia repair
Definition
Esophageal atresia repair, also known as tracheoesophageal fistula or TEF repair, is a surgical procedure performed to correct congenital defects of the esophagus (the muscular tube that connects the mouth to the stomach) and the trachea (the windpipe that carries air into the lungs). Esophageal atresia (EA) and tracheoesophageal fistula (TEF) are commonly found together (EA/TEF), but may also occur separately. As of 2003, there is no known cause for these congenital defects.
Purpose
In children born with EA, the esophagus has not developed as a continuous passage into the stomach but ends in a blind pouch. In the majority of cases (86%) it is also abnormally connected to the trachea by a small channel called a fistula. EA/TEF repair is performed to correct these defects, ensuring the survival of affected infants and their proper breathing and digestion.
Demographics
EA/TEF is reported to occur in about 1: 4500 births. It occurs equally among male and female infants and has been associated with prematurity. There are no other notable associations.
Description
The human esophagus and trachea are normally formed as two separate but parallel passageways early in fetal development. The esophagus leads from the throat to the stomach and digestive tract, and the trachea leads from the larynx to the lungs and respiratory system. Esophageal atresia occurs when the esophagus is incompletely formed; most typically, its upper portion ends in a pouch, failing to connect with the lower portion that leads to the stomach. Esophageal atresia with tracheoesophageal fistula, commonly known as EA/TEF, occurs when the membrane that divides the trachea from the esophagus (tracheoesophageal septum) is incompletely
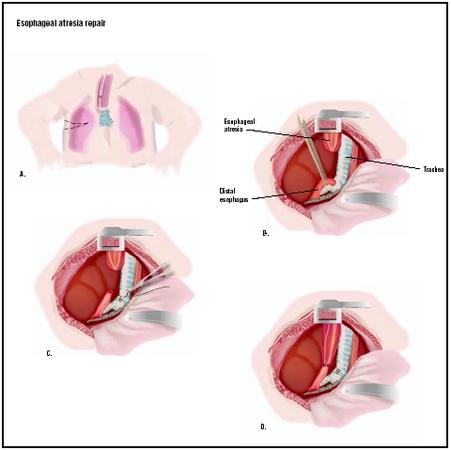
A classification system is commonly used to describe five types of esophageal atresia, with and without TEF:
- Type A: Esophageal atresia (7.7% of cases). EA alone is a condition in which both segments of the esophagus, upper and lower, end in blind pouches with neither segment attached to the trachea.
- Type B: Esophageal atresia with tracheoesophageal fistula (86.5%). This is the most common type of EA/TEF, in which the upper portion of the esophagus ends in a blind pouch and the lower segment of the esophagus is attached to the trachea by a fistula.
- Type C: Esophageal atresia with tracheoesophageal fistula (0.8%). Type C is a rare form of EA/TEF in which the upper segment of the esophagus forms a channel to the trachea (TEF) and the lower segment of the esophagus ends in a blind pouch (EA).
- Type D: Esophageal atresia with tracheoesophageal fistula (0.7%). Type D is the rarest form of EA/TEF, in which both segments of the esophagus are attached to the trachea.
- Type H: Tracheoesophageal fistula (4.2%). TEF alone is a condition in which a fistula is present between the esophagus and the trachea, while the esophagus has a normal connection to the stomach.
The incomplete esophagus in EA/TEF will not allow swallowed saliva, food, or liquids to pass into the stomach for normal digestion and nutrition. Because of the defect, normal eating or drinking can be dangerous because food and fluids have a direct route through the fistula into the lungs. Swallowed material in the dead-end esophagus as well as stomach fluids may be aspirated into the lungs through the fistula, compromising the child's breathing and potentially causing pneumonia or infection. The impossibility of normal eating, breathing, and digestion creates a life-threatening condition that requires immediate surgery.
The first signs of EA/TEF in a newborn infant may be tiny white frothy bubbles of mucus in the infant's mouth and sometimes in the nose as well. These bubbles reappear when they are suctioned away. Although the infant can swallow normally, the parents can often hear a rattling sound in the chest along with coughing and choking, especially when the baby is trying to nurse. Depending on the severity of the defect, some infants may develop a bluish complexion (cyanosis), caused by a lack of sufficient oxygen in the circulatory system. The infant's abdomen may be distended (swollen and firm) because the abnormally formed trachea will allow air to build up in the stomach and fill the space surrounding the abdominal organs. Saliva and stomach fluids may be aspirated into the lungs through the abnormal opening in the infant's trachea. Aspiration can lead to infection or even asphyxiation (impaired breathing or loss of consciousness due to lack of oxygen).
Other congenital defects are found in at least 50% of infants with EA/TEF. Typically more than one type of malformation will be found. These may include:
- Heart defects (about 25% of affected infants)
- Gastrointestinal (digestive) anomalies, including malformed anus (rectum) or twisting of the small intestine (about 16%)
- Urinary tract and kidney defects (10%)
- Musculoskeletal (muscle and bone) defects, especially of the ribs and limbs
The multiple anomalies that can occur with EA/TEF have been described by an acronym, VATER or VACTRRL. This acronym stands for v ertebral defect, a norectal malformation, c ardiac defect, t racheoesophageal fistula, r enal anomaly, r adial dysplasia, and l imb defects. About 10% of children with EA have what is called the VATER syndrome. More infants with Type A esophageal atresia have multiple anomalies than those with Type B, the combined EA/TEF.
Healthy infants who have no complications, such as heart or lung problems or other types of intestinal malformations, can usually have esophageal surgery within the first 24 hours of life. The operation will be delayed for low birth weight infants or those with complicated malformations, usually until their nutritional status can be improved and other problems resolved sufficiently to reduce the risks of surgery. H-type TEF, which has fewer symptoms and is typically diagnosed when the child is at least four months old, is also easier to repair when the child's size and weight have increased. The esophagus can be dilated periodically during the growth period and a stomach tube used to decompress the stomach until the surgical repair is performed. All infants with some type of esophageal atresia will require surgery; many will have the repair performed in separate stages over a period of years. The procedures used to treat the five types of EA/TEF defects are similar.
Surgery is conducted while the infant is under general anesthesia, unconscious and free of pain. The surgeon makes an incision in the right chest wall between the ribs. If the gap between the two portions of the esophagus is short, the surgeon may join both ends of the esophagus together. This is called an anastomosis. If the upper portion of the esophagus is short and a long gap exists between the upper and lower portions, reconstructive surgery cannot be performed and the infant will have to be fed in another way to allow several months of growth. In this case, a gastrostomy (stomach tube) may be surgically placed directly into the stomach for feeding. In the most typical EA/TEF repair, the fistula will first be closed off, creating a separate airway. Then the blind esophageal pouch will be opened and connected to the other portion of the esophagus, creating a normal pathway directly into the stomach.
Diagnosis/Preparation
Diagnosis
A tentative diagnosis of EA/TEF may be made before the child is born. One of the first signs of esophageal atresia may be seen during the mother's prenatal ultrasound examination. Polyhydramnios, which is an excessive amount of amniotic fluid surrounding the fetus, is not always diagnostic but offers a warning sign. Fluid is normally exchanged between the fetus and the amniotic fluid through swallowing, urination, and discharges from the nose and mouth. In EA/TEF, the fetus may drool excessively because of a collection of fluid in the abnormal esophageal pouch, thus increasing the amount of amniotic fluid.
A newborn infant suspected of EA/TEF will be given an x-ray examination. The imaging study may reveal a dilated esophageal pouch that is larger than expected because it has collected a pool of amniotic fluid. During fetal development, the enlarged esophagus may also have pressed on and narrowed the trachea, a condition that may contribute to fistula development. Air in the stomach may confirm the presence of a fistula while gas in the large intestine will rule out duodenal atresia. The physician will also perform a comprehensive physical examination , looking for other congenital anomalies that are known to accompany EA/TEF. Chest x-rays may be taken to look for skeletal and cardiac abnormalities. Abdominal X-rays may be taken as well to look for intestinal obstruction and abnormalities. An echocardiogram (ECG) may be performed to evaluate heart function and an ultrasound of the kidneys performed to evaluate kidney function. A nasogastric (NG) tube may be placed in the infant; it may help to confirm a diagnosis of EA/TEF if it stops short of the usual distance (17 cm or about 6.7 in) to the stomach.
Preparation
When an infant is suspected of having EA/TEF, he or she will be transferred from the regular nursery to the neonatal (newborn) or pediatric intensive care department of the medical facility. Corrective surgery must be scheduled immediately to help ensure survival and promote proper swallowing, digestion, nutrition, and breathing. In patients with pneumonia or other lung problems, the doctor may clean out the baby's stomach and esophageal pouch with a suction tube to prevent the baby's stomach contents from being drawn through the fistula and into the trachea. A tube will be placed through the baby's mouth to continuously suction the esophageal pouch during surgery. The baby will be given fluids intravenously during surgery. Oxygen therapy will be administered if needed. An airway will be placed in the trachea if the baby has lung problems. Antibiotics may be given to treat or to prevent infection in the lungs, especially if the stomach contents have been drawn into the lungs. Preoperative blood and urine tests will also be performed.
Aftercare
Immediately after surgery, the patient will be cared for in the neonatal ICU with monitoring of breathing, body temperature, and heart and kidney function. Oxygen may be administered, and a mechanical respirator may also be necessary. Pain medication will be given if needed. Blood and urine tests may be performed to evaluate the infant's overall condition. Scans may be performed to evaluate esophageal functioning. The infant will be fed intravenously or will have a gastrostomy tube placed directly into the stomach until oral feedings can be swallowed and digested. Secretions may be suctioned from the throat and a nasogastric tube may be placed in the infant's nose to clear the stomach as needed. Hospitalization may be required for two weeks or longer, depending on the presence of complications or other underlying conditions. An x-ray procedure known as esophagography is usually performed at two months, six months, and one year of age to monitor the digestive function as the child grows. Long-term follow-up of patients who have had EA/TEF repair is essential.
Risks
There is some risk of postoperative breathing difficulties or respiratory complications, particularly infection or pneumonia due to aspiration of stomach contents. Pretreatment with antibiotics helps to prevent infection to some degree. Other risks of esophageal atresia repair include those that may occur with any surgery: reactions to anesthesia or medications; bleeding or clot formation; narrowing of the repaired organs; nerve injury; fluid imbalances; and collapsed lung (pneumothorax).
Complications that can occur in infants who have had EA/TEF repair include the following:
- Esophageal dysmotility. Dysmotility refers to weakness of the muscular walls of the esophagus. Some degree of dysmotility is expected to occur in all infants undergoing esophageal repair. This complication requires special precautions when the child is eating or drinking.
- About 50% of patients will develop gastroesophageal reflux disease (GERD) later in childhood or adult life. GERD is a condition in which the acid contents of the stomach flow back into the esophagus. The condition requires medical or surgical treatment.
- Recurrence of TEF. Recurrence is treated with repeat surgery.
- Swallowing difficulties (dysphagia). Dysphagia can cause food or pills to become stuck in the esophagus at the site of the surgical repair. Medications should be taken with water to prevent ulcers from developing.
- Breathing difficulties and choking. These complications are related to the slow passage of food, food stuck in the esophagus, or aspiration of food into the trachea.
- Chronic cough. The cough is characteristic of TEF repair; it is caused by weakness in the trachea and does not indicate a cold or illness.
- Increased susceptibility to colds, respiratory infections, and pneumonia. Precautions should be taken to avoid contact with other sick children. Parents and caregivers can seek advice about strengthening the child's immune system through appropriate nutrition and supplements.
Normal results
EA/TEF can usually be corrected with surgery, allowing the child to eat, breathe, and digest food in a normal fashion. Although almost 100% of children who have corrective surgery for EA/TEF survive the procedure, they may continue to have complications, some of which can be chronic. Ongoing medical care and additional surgery may be necessary.
Morbidity and mortality rates
The post-operative mortality rate in healthy infants is essentially zero. Prior to the development of a more advanced EA/TEF repair technique in 1939, the condition was often fatal. Pre- and postoperative neonatal care has also significantly improved the surgical outcome. Infants with multiple anomalies or cardiac or pulmonary problems are more subject to complications than otherwise healthy infants.
Alternatives
Esophageal atresia and tracheoesophageal fistula are congenital defects for which there are no recommended alternatives. These defects are commonly corrected surgically. There are no known measures to prevent these congenital defects.
Resources
books
Allan, W., MD, et al. Pediatric Gastrointestinal Disease: Pathophysiology, Diagnosis, Management , 3rd ed. Boston, MA: B.C. Decker, 2000.
organizations
American Academy of Family Physicians. PO Box 11210, Shawnee Mission, KS 66207. (800) 274-2237. http://www.aafp.org .
EA/TEF Child and Family Support Connection. 111 West Jackson Blvd., Suite 1145, Chicago, IL 60604. (312) 987-9085. http://www.eatef.org .
other
Allesandro, M. P., MD. Esophageal Atresia and Tracheoesophageal Fistula (EA/TEF) . Gastrointestinal Diseases, Virtual Children's Hospital Library. http://www.vh.org/pediatric/provider/radiology/PAP/GIDiseases/EATEF.html .
TEF/VATER International. What is esophageal atresia? http://www.tefvater.org .
L. Lee Culvert
WHO PERFORMS THE PROCEDURE AND WHERE IS IT PERFORMED?
EA/TEF repair is performed in a hospital operating room by a pediatric or general surgeon.
QUESTIONS TO ASK THE DOCTOR
- Is surgery the only solution? When will surgery be performed and what will be the likely results?
- How many children have you treated with this procedure? What were the results?
- Will my child be able to breast feed or drink milk from a bottle? How soon after surgery?
- How soon can I take my child home?
- What symptoms should I watch for and what kind of problems may occur when we get home?
- How will my child's early years be affected by this condition and the repair surgery? Will my child enjoy normal eating, growth, and social activities?
- What are the chances of my child needing another operation?
Please send me an answer.
I had dilation at 6 mths, 2 yrs, and 6 yrs due to stricture.
I am now 36 y. old with GERD--but take daily Prilosec to suppress the acid.
Clearly avoid any large objects in respect to your baby--swallowing is still a problem.
I also have loss of motility.
Other than that, I am fine.
Any specifics, email me at cpaver1@hotmail.com
Cheers
MY SON IS HAVING A PROBLEM BY BIRTH OF TEF .3 TIMES ULTRASOUND WAS DONE OF MY WIFE BEFORE HIS BIRTH .CAN I CLAIM ON THE DOCTOR WHO WAS DOING TREATMENT OF MY WIFE ?
AS IT COST VERY MUCH TO OPERATE IT AS I AM WORKING AS A WORKER IN PRIVATE COMPANY AND I CANT'T AFFORD IT SO PLS ADVICE ME FOR THAT.
WITH REGARDS
YOGESH
For those with infants facing this, have faith. He's 52, a father and successful.
My Dad is 74 but wondered if anyone had some reassuring information.
He had his hole in oesphagus repaired but there was still a leak. They thought it had repaired itself as the drain was empty.
Then he had vomiting and there is small fluid in the drain. Does this mean maybe he has another tear in the oesphagus?
my daughter is now 8 years old. she was born TEF - corrected in 11 months. I live in chennai. she has been operated and tread by the ICH- Institue of child health, Egmore chennai. The hospital is well equipped to handle birth anomalies with highly qualified dedicated doctors. I am trying to form a support group for TEF in chennai. Treating and living with children of TEF is a life time commitment. A little help, support and comforting words help the family and the child to handle the problem well. any one interested in joining us u are welcome( it involves no fee/charges only love)
kavitha
I'm working with a 3 years old girl who was born with Tracheo-Oesophageal-Fistula (TOF) and Oesophageal Artresia.
She had a repair operation in April 2010 resulting in the reposition of her stomach into her chest. Now jejostomy fed (fed into her jejunum and not her stomach)
Has been having episodes of sickness since she was 'joined up' in April 2010.
Sickness was about 4 weeks apart but are now 2-3 weeks in between and last about 3-4 days, where she wretches up saliva and bile and then sleeps.
Is there somebody who could givew me some advise what could cause this problem or how to sort out? I'm really apprecite for your help.
Kind regards,
Barbara
Have faith, your child will get past these hurdles and be a stronger person.
I at times i cough and cough. Apparently it was sort of a rare type surgery the doctors performed but it has seemed to work successfully over the year for me. I can't really digest beef steak or pork as its way too heavy for my stomach to digest properly. I tend to eat fish, processed meats as its easier for me to swallow and digest. I use the bathroom normally. It's just simply amazing the technology doctors really had back in the 50's. I'm 54 now and still going strong. I do not smoke which is a good thing. My advice to everyone who ever had this surgery done. It's worth it. Believe me if i hadn't had this surgery i probably wouldn't be here today telling you this. God bless the doctors and nurses that made it all happen.
My case involved a total disconnect of the esophogaus and a blind pouch which forced any liquid or food intake directly to my lungs. After a few days from my birth the doctors discovered the condition and I was rushed to NY for the procedure. My informtion has it that I was one of the first to have a procedure that did not involve any intestinal grafting but instead a repositioning that permitted the reconnection of the two distal ends of my esophogaus and removal of the pouch. The procudre was written up in the New England Medical Journals. I do have to be carefull to take small bites of food as to not have them become stuck, on what I call the ledge in my throat, but other than tha I have not had any other bad affects. I thank God for all who helped me back in the day.
I was lucky and bless to have great doctors and nurses and Parents who let me grow up and do all the sports etc as any other kid
It was kept a secret because I was adopted at birth & my adopted parents paid for the operation.
Regards
Narelle Mason